
Carrier Screening Guide: How to Ensure Reliable Information for All Patients
Updated information on cystic fibrosis, spinal muscular atrophy, and fragile X syndrome carrier screening for clinicians.
Overview
Advanced Genetic Testing Delivers More Reliable Results for Carrier Screening
Three of the conditions most frequently recommended for carrier screening are cystic fibrosis
(CF), spinal muscular atrophy (SMA), and fragile X syndrome (FXS). Unfortunately, conventional
approaches have tended to deliver uneven results across ancestries, and they often fail to detect
more complex genetic variants with important clinical implications.
Recently, the American College of Medical Genetics and Genomics (ACMG) updated its carrier
screening guidelines1. The new recommendations emphasize the importance of adopting methods
that produce meaningful results for patients of all ancestries — and of allowing physicians to make
consistent carrier screening recommendations for all patients without having to base decisions on
ancestry or ethnicity.
Carrier screening is moving to a pan-ancestry model. This transition will offer enormous benefit to patients by ensuring more reliable results for all. However, getting there will require abandoning genetic tests that don’t hold up to this new standard, and ensuring that all carrier screening tests utilized are based on insights gleaned from diverse, population-scale data sets. In addition, these tests must be robust enough to reliably detect even the most challenging genetic variants.
population-scale data sets. In addition, these tests must be robust enough to reliably detect even the most challenging genetic variants.
How does this affect carrier screening for commonly tested conditions including cystic fibrosis, spinal muscular atrophy and fragile X?
Consider these important facts related to carrier screening for each disease:
Cystic fibrosis
- 95% of cystic fibrosis variant information in the “gold-standard” historical database is from
people of European descent2 - Multiple studies3,4 have found the ACMG 23-variant recommendation to be insufficient for
detecting cystic fibrosis carriers in diverse populations - More variants do not guarantee more coverage: larger variant panels still lack sufficient
coverage of diverse populations because they were designed based on ethnically
homogeneous data3,5 - People of non-European descent are less likely to be detected on prenatal and newborn
screening tests even when they are carriers or have cystic fibrosis, due to use of panels lacking
coverage for diverse populations6,7,8
Spinal Muscular Atrophy
- If a test is reporting SMN1 copy number alone for carrier screening, there can be up to
approximately 30% false negative results, depending on patient ancestry9 - These false-negative results are due to “silent carriers” that have 2 copies of the SMN1 gene,
but both copies of the gene are on the same allele. Thus, a “silent carrier” can still pass on an
allele with no copies of SMN1 in this case. If both parents pass on alleles with no copies of
SMN1, the child will have SMA. - This “silent carrier” phenomenon is more common in people of African descent9, meaning
determining the risk of “silent carrier” genotype may be very important in this demographic
Fragile X Syndrome
- To determine the risk of a potential carrier having a child with fragile X, triplet repeats (CGGs)
in the FMR1 gene must be quantified. However, it is difficult for many technologies to quantify
these repeats accurately. - In addition, there are triplet-repeat interrupting sequences (AGGs) that can significantly alter
the risk of certain women for having a child with Fragile X4,10,11. For example, quantifying AGG
interruptions adjusts the risk for 90% of certain female carriers compared to CGG repeats
alone4, but clinicians may not be receiving this information for their patients.
Fortunately, the public release of data from diverse large-scale genomic studies has now made
it possible to identify relevant genetic alterations across populations. By incorporating this
information into carrier screening tests, and by building these tests with advanced technology
capable of detecting all relevant genetic information regardless of complexity, modern carrier
screening can deliver reliable results for all patients.
Cystic Fibrosis Carrier Screening
Underrepresented genetic diversity in data sets is a common challenge in genomics today. The
effects of this can be seen in diagnostic and carrier screening tools that provide more accurate and
comprehensive results for some populations than for others.
Consider these metrics about available genomic data sets: In 2009, an analysis of genome-wide
association studies found that 96% of the 1.7 million samples reviewed came from individuals of
European ancestry12. By 2021, data from the GWAS Diversity Monitor website13 indicated that the
share of GWAS samples from people with non-European ancestry had grown from that measly 4%
to just 12%.
The vast majority of historically available data supporting cystic fibrosis testing also comes from
people of European descent. The prominent historical database2 includes data from nearly 90,000
patients on pathogenic variants detected in the CFTR gene, connecting patient genotype and
phenotype. It has long been considered the gold standard on genetic data for cystic fibrosis.
Unfortunately, of the patients represented in the database, 95% have European ancestry, leaving
other populations woefully underrepresented (Figure 1).

Figure 1. Illustration of the historical variant database2 patient ethnicity compared to more realistic representation of U.S. population diversity3.
To add to the challenges in CFTR testing, there have been over 2000 CFTR mutations documented
14, with varying levels of pathogenicity. In order to provide practical cystic fibrosis
carrier screening guidance for clinical laboratories, ACMG recommended testing for 23 of the
most common mutations in 2004, based on available data at that time15.
 Since then, several large-scale studies have reported the lack of coverage for diverse populations by the ACMG 23 recommended variants. One study of over 370,000 people with an ethnic distribution representative of the U.S. population found that 44% of carriers would have been missed using the ACMG 23 panel4. Another recent study of over 13,000 screened
Since then, several large-scale studies have reported the lack of coverage for diverse populations by the ACMG 23 recommended variants. One study of over 370,000 people with an ethnic distribution representative of the U.S. population found that 44% of carriers would have been missed using the ACMG 23 panel4. Another recent study of over 13,000 screened
couples with an ethnic distribution representative of the U.S. population found that 31% of at-risk couples would have been undetected using the ACMG-23, which would have resulted in missed cases of cystic fibrosis3.
Even more concerning is that most currently used targeted cystic fibrosis screening panels are at least five years old and were designed based on the historical database. Though these panels contain many more variants than the ACMG-23 panel, they still lack sufficient carrier screening coverage for the diverse U.S. population (Table 1). This is due to their design based on outdated and ethnically homogeneous data. The lack of coverage for specific ethnicities can further be seen for an on-market panel in Table 2.
Table 1. Three on-market CFTR panels with more variants detected than ACMG 23, but lacking sufficient cystic fibrosis carrier detection for the diverse U.S. population.

Table 2. Manufacturer-reported CFTR mutation detection rates of a commonly used CFTR targeted panel5. Reported coverage percentages are based on data published in 2004 or earlier, meaning coverage could be poorer if compared with more recently published, diverse datasets.

However, universal carrier screening is recommended for cystic fibrosis, which means clinicians need results that accurately represent the risk of passing on this disease for patients of all ancestries.
Large-scale population studies are adding rapidly to the information available about pathogenic variants associated with cystic fibrosis in people of diverse genetic backgrounds. Some of these studies deliberately gather data from diverse populations, such as that of the United States3,4. The rush of new, publicly-available genomic data represents significant progress in identifying relevant variants across ancestries.
Still, this information must be incorporated into carrier screening tests to make a difference for prospective parents wondering if their children would be at risk of inheriting cystic fibrosis. Clinicians ordering carrier screening tests for cystic fibrosis should reach out to their clinical lab teams to ensure that such tests have been recently updated to reflect more diverse data sources — covering not only the most common variants found in the historical database but also diverse population-specific variants identified in large-scale sequencing studies.
Spinal Muscular Atrophy (SMA) Carrier Screening
Until recently, a diagnosis of spinal muscular atrophy (SMA) was considered a death sentence. This debilitating neuromuscular disease is a leading genetic cause of death among infants. The introduction of game-changing therapies in the last few years, though, has brought new hope to families of affected individuals16.
Though these therapies have been revolutionary, they do not negate the need for carrier screening. In fact, SMA carrier screening is recommended by the American College of Obstetricians and Gynecologists (ACOG) and ACMG for all women who are pregnant or planning to become pregnant1,17. SMA has a high carrier rate of approximately 1 in 50 individuals18.
In addition, SMA screening accuracy can vary depending on ancestry. Historically, SMN1 copy number was thought to be the only information needed for SMA carrier status: if a person had two copies of SMN1 it was assumed they were on different alleles and the person could not pass on the disease to a child. Now it is known that conventional tests that report only SMN1 copy number may miss “silent carriers”, or people who carry two copies of the SMN1 gene on a single allele and no copies on the other allele (Figure 2). This silent carrier genotype can occur in any ethnicity, but it is more common among people of African ancestry9, so standard SMA screening tests may be more likely to miss carriers in this population. In fact, if reporting SMN1 copy number alone, “silent carrier” genotypes can lead to approximately 30% false negative results for African American patients9.
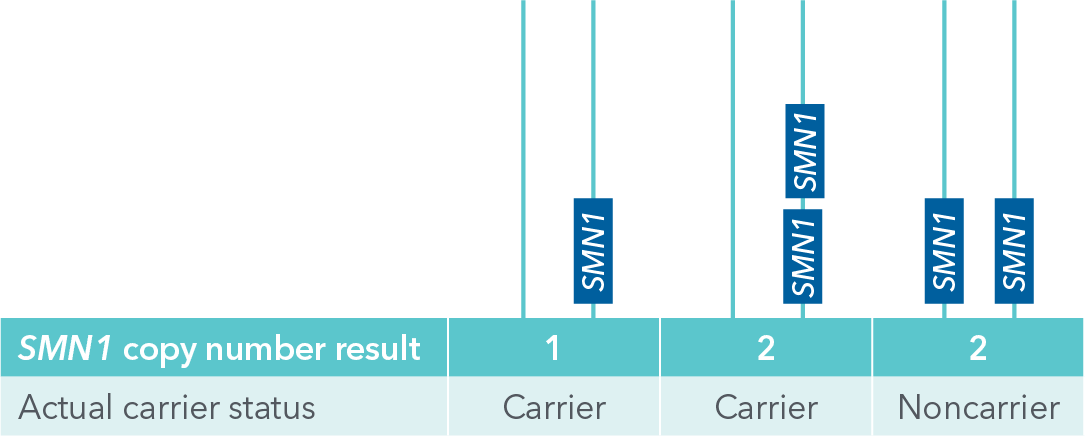
Figure 2. Illustration of SMN1 “silent carrier” genotype with two SMN1 copies on the same allele (center), which can allow for passing on a zero copy allele, compared to a noncarrier genotype where one SMN1 copy is present on each allele (right). One copy SMN1 carrier genotype is on left. If receiving SMN1 copy number results only, silent carriers cannot be distinguished since both center and right genotypes would yield results of 2 copies of SMN1.
Fortunately, it has been observed that certain variants (called “linked variants”) in the SMN1 gene can help predict silent carrier status. Presence of these variants can help further stratify carrier risk in all ethnicities, and even diagnose carrier status in a few19, 20 (Table 3).
Table 3. Impact of positive results for an SMN1 linked variant (c.*3+80T>G) on SMA carrier risk19, 20 when positive for two SMN1 copies, by ethnicity.

It is no longer enough to report only SMN1 copy number for carrier screening. All SMA carrier screening tests should also include detection of these linked variants to provide patients with better information on potential carrier status.
Furthermore, many commonly used tests for SMA are not as accurate or as sensitive as they should be when providing such vital clinical information. The test considered the gold standard for SMA testing is based on a cumbersome, time-consuming technology called MLPA. There has been some concern cited that this technology many not reliably distinguish certain copy numbers21. In addition, popular SMA screening technologies may not provide linked variant detection to inform carrier risk. Clinicians should check with their clinical lab teams to ensure they are using an SMA carrier screening test capable of providing this information.
Fragile X Carrier Screening
Fragile X syndrome is the most common inherited form of intellectual disability and autism, affecting approximately 1 in 4,000 males and 1 in 5,000 females in the United States22. ACOG and ACMG recommend carrier screening for this syndrome1,17,23. However, of the estimated 1 million women believed to be Fragile X carriers in the U.S.22, the majority may be unaware of their carrier status.
Fragile X syndrome is caused by a CGG repeat expansion in the FMR1 gene on the X chromosome. Different counts of the repeat have different phenotypic outcomes (Table 4). Healthy individuals typically have fewer than 45 CGG repeats, while those who may be unaffected carriers or have associated disorders such as Fragile X-Associated Primary Ovarian Insufficiency (FXPOI) generally have between 55 and 200 repeats. Individuals with at least 200 repeats are diagnosed with Fragile X syndrome.
Table 4. Clinical categories and carrier status of mutations in the FMR1 gene24,25,26.
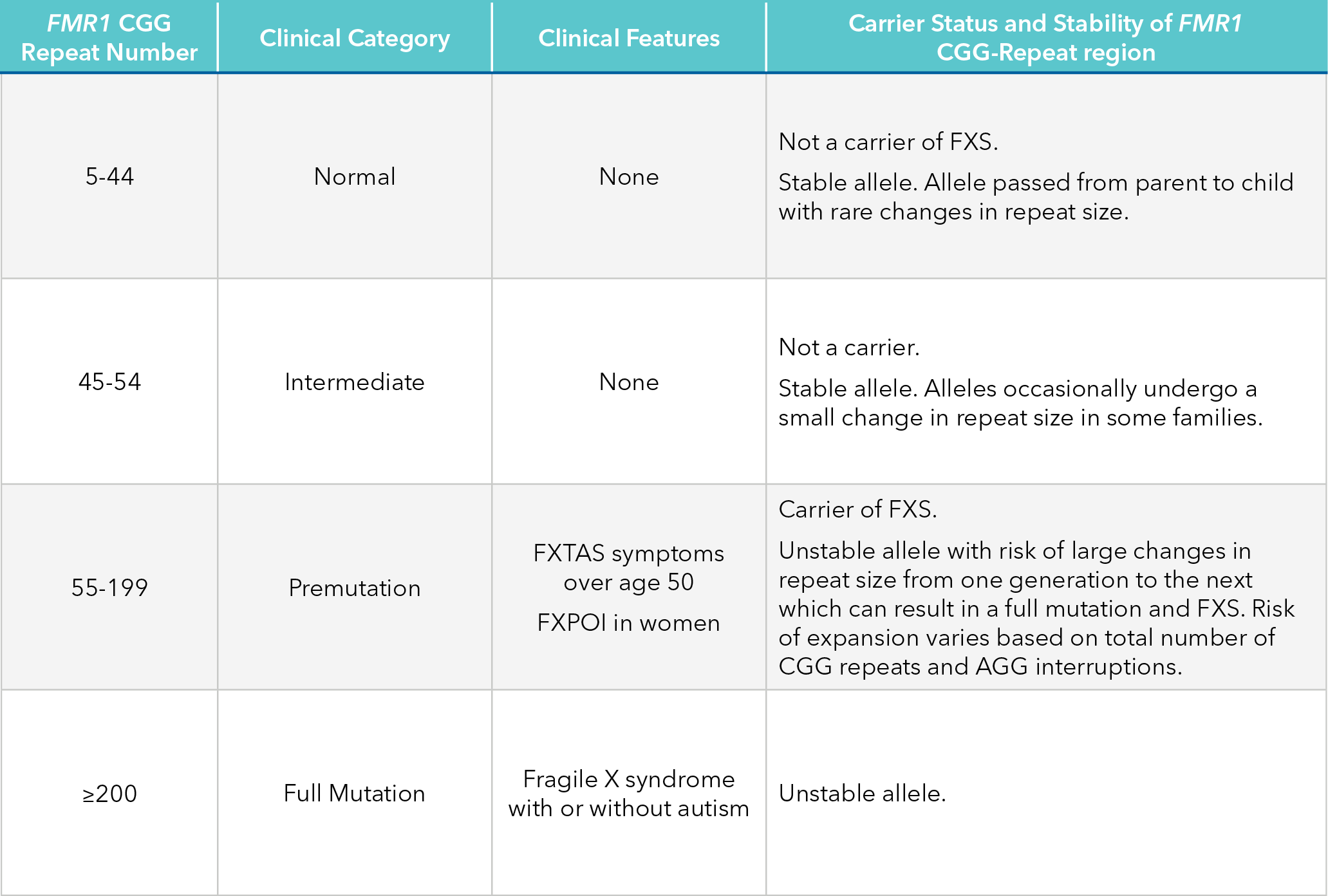
Abbreviations: FXS, fragile X syndrome; FXTAS, fragile X-associated tremor/ataxia syndrome; FXPOI, fragile X-associated primary ovarian insufficiency.
For any genetic testing platform, obtaining an accurate count of so many repeated DNA triplets is a serious challenge. Additionally, many analysis platforms cannot accurately interrogate the region due to the technical challenges of assessing GC-rich DNA content. Therefore, it is important that fragile X carrier screening is being performed with robust technology that can analyze extreme GC content and provide accurate counts of CGG repeats in order to identify women at risk of having a child with fragile X.
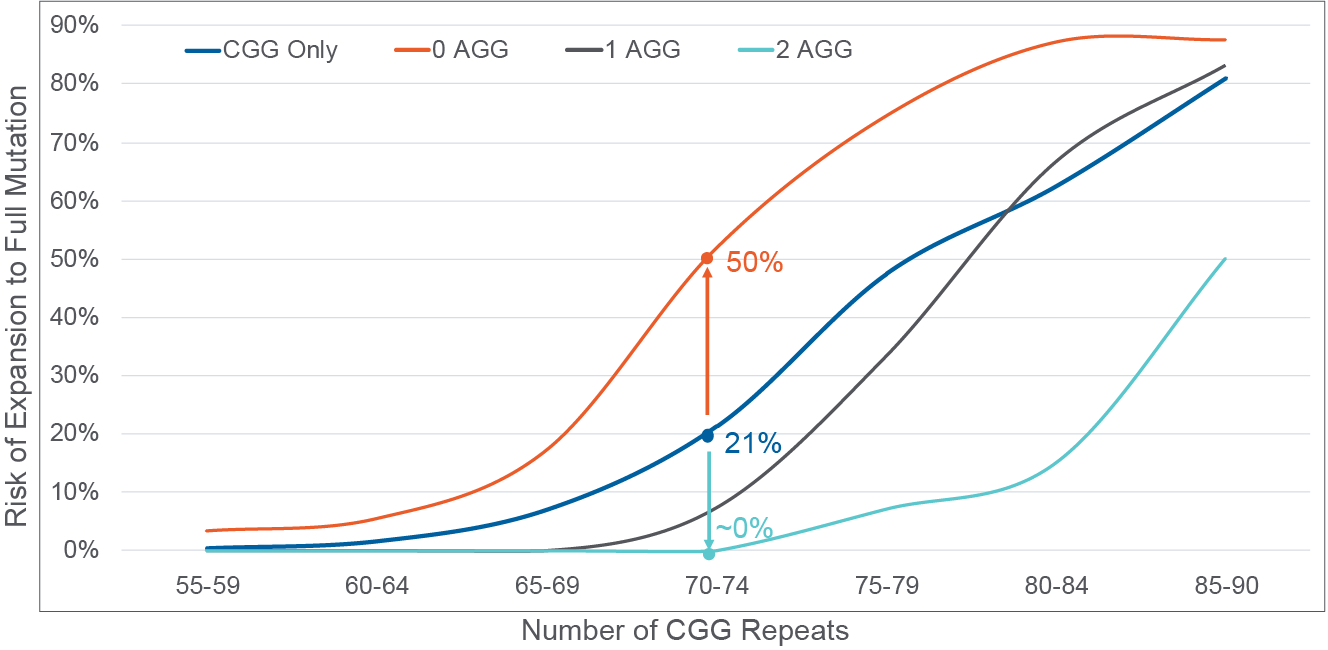
Figure 3. AGG interruption quantification stratifies risk of expansion to full mutation in the next generation10,11,27. Risk by CGG repeat number only represented by blue line; 0 AGG interruptions detected, orange line; 1 AGG interruption detected, black line; 2 AGG interruptions detected, teal line. A 70 CGG patient example is illustrated where 21% risk is reported with CGG repeat-only information, which can be further stratified to range from 50% risk when 0 AGG interruptions (orange arrow) are detected to near 0% risk when 2 AGG interruptions are detected (teal arrow).
Beyond CGG repeat counts, clinicians should also be provided information on AGG interruptions for women with 50 – 90 CGG repeats. In this select group, the number of AGG interruptions stratifies risk of expansion to a full mutation in offspring beyond that provided by CGG repeats alone (Figure 3). For example, for a woman with a 70 CGG repeat allele, risk of expansion to a full mutation can range from ~0% to 50% depending on the number of AGG interruptions. Information provided by the AGG interruption status can greatly affect reproductive decisions; thus, clinicians should work with their clinical lab partners to ensure they are providing this information for select patients.
Conclusion
As insight into inherited diseases grows and diagnostic technology advances, providing all patients with accurate and relevant information must remain the central goal. Thankfully, due to the diligence of many scientists and clinicians, better tests have been developed for cystic fibrosis, spinal muscular atrophy, and fragile X carrier screening. As guidelines have shifted to pan-ethnic carrier screening, it is vital that clinicians and their clinical lab partners are aware of disparities and gaps in genetic information from often-used, yet outdated tests, and that they strive to provide more comprehensive and reliable information for all patients.
References
1. Gregg AR, et al. Genet Med. 2021 Oct;23(10):1793-1806.
2. Sosnay et al Nat Genet. 2013 Oct; 45(10): 1160–1167
3. Beauchamp KA, et al. Genet Med. 2019; 21(11): 2569–2576.
4. Westemeyer M, et al. Genet Med. 2020 Aug;22(8):1320-1328.
5. Luminex xTAG® Cystic Fibrosis (CFTR) 60 kit v2 product brochure.
6. McGarry ME, et al. Pediatr Pulmonol. 2019 Nov;54 (Suppl 3):S74-S83.
7. Watts KD, et al. J Genet Couns. 2012 Oct;21(5):671-5. doi: 10.1007/s10897-012-9481-2.
8. Neemuchwala F, et al. Case Rep Pediat. 2018 Jul 2;2018:7217326.
9. Hendrickson BC, et al. J Med Genet. 2009 Sep;46(9):641-4.
10. Nolin SL, et al. Genet Med. 2015 May;17(5):358-64.
11. Nolan SL, et al. Am J Med Genet A. 2013 Apr;161A(4):771-8.
12. Need AC and Goldstein DB. Trends Genet. 2009 Nov;25(11):489-94.
13. Mills MC and Rahal C. GWAS Diversity Monitor. 2021. https://gwasdiversitymonitor.com/
14. Veit G, et al. Mol Biol Cell. 2016 Feb 1; 27(3): 424–433.
15. Watson MS, et al. Genet Med. Sep-Oct 2004;6(5):387-91.
16. Nance JR. Spinal Muscular Atrophy. Continuum (Minneap Minn). 2020 Oct;26(5):1348-1368.
17. Rink B, et al. ACOG Opinion No 691. March 2017. Reaffirmed 2020.
18. Cure SMA. 2021. https://www.curesma.org/carriers-of-sma/
19. Luo, et al. Genet Med. 2014 Feb;16(2):149-56.
20. Alías L, et al. Eur J Hum Genet. 2018 Oct;26(10):1554-1557.
21. Schorling DC, et al. Neurology. 2019 Aug 6;93(6):267-269.
22. National Fragile X Foundation. 2021. https://fragilex.org/
23. Sherman S, et al. Genet Med. 2005 Oct;7(8):584-7.
24. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 300623. 2019. https://omim.org/
25. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 300624. 2019. https://omim.org/
26. Allen EG, et al. Genet Med. 2021 Sep;23(9):1648-1655.
27. Villate O, et al. Front Mol Biosci. 2020 Jul 14;7:135.
Carrier Screening Guide: How to Ensure Reliable Information for All Patients
Updated information on cystic fibrosis, spinal muscular atrophy, and fragile X syndrome carrier screening for clinicians.
Overview
Advanced Genetic Testing Delivers More Reliable Results for Carrier Screening
Three of the conditions most frequently recommended for carrier screening are cystic fibrosis
(CF), spinal muscular atrophy (SMA), and fragile X syndrome (FXS). Unfortunately, conventional
approaches have tended to deliver uneven results across ancestries, and they often fail to detect
more complex genetic variants with important clinical implications.
Recently, the American College of Medical Genetics and Genomics (ACMG) updated its carrier
screening guidelines1. The new recommendations emphasize the importance of adopting methods
that produce meaningful results for patients of all ancestries — and of allowing physicians to make
consistent carrier screening recommendations for all patients without having to base decisions on
ancestry or ethnicity.
Carrier screening is moving to a pan-ancestry model. This transition will offer enormous benefit to patients by ensuring more reliable results for all. However, getting there will require abandoning genetic tests that don’t hold up to this new standard, and ensuring that all carrier screening tests utilized are based on insights gleaned from diverse, population-scale data sets. In addition, these tests must be robust enough to reliably detect even the most challenging genetic variants.
How does this affect carrier screening for commonly tested conditions including cystic fibrosis, spinal muscular atrophy and fragile X?
Consider these important facts related to carrier screening for each disease:
Cystic fibrosis
- 95% of cystic fibrosis variant information in the “gold-standard” historical database is from
people of European descent2 - Multiple studies3,4 have found the ACMG 23-variant recommendation to be insufficient for
detecting cystic fibrosis carriers in diverse populations - More variants do not guarantee more coverage: larger variant panels still lack sufficient
coverage of diverse populations because they were designed based on ethnically
homogeneous data3,5 - People of non-European descent are less likely to be detected on prenatal and newborn
screening tests even when they are carriers or have cystic fibrosis, due to use of panels lacking
coverage for diverse populations6,7,8
Spinal Muscular Atrophy
- If a test is reporting SMN1 copy number alone for carrier screening, there can be up to
approximately 30% false negative results, depending on patient ancestry9 - These false-negative results are due to “silent carriers” that have 2 copies of the SMN1 gene,
but both copies of the gene are on the same allele. Thus, a “silent carrier” can still pass on an
allele with no copies of SMN1 in this case. If both parents pass on alleles with no copies of
SMN1, the child will have SMA. - This “silent carrier” phenomenon is more common in people of African descent9, meaning
determining the risk of “silent carrier” genotype may be very important in this demographic
Fragile X Syndrome
- To determine the risk of a potential carrier having a child with fragile X, triplet repeats (CGGs)
in the FMR1 gene must be quantified. However, it is difficult for many technologies to quantify
these repeats accurately. - In addition, there are triplet-repeat interrupting sequences (AGGs) that can significantly alter
the risk of certain women for having a child with Fragile X4,10,11. For example, quantifying AGG
interruptions adjusts the risk for 90% of certain female carriers compared to CGG repeats
alone4, but clinicians may not be receiving this information for their patients.
Fortunately, the public release of data from diverse large-scale genomic studies has now made
it possible to identify relevant genetic alterations across populations. By incorporating this
information into carrier screening tests, and by building these tests with advanced technology
capable of detecting all relevant genetic information regardless of complexity, modern carrier
screening can deliver reliable results for all patients.
Cystic Fibrosis Carrier Screening
Underrepresented genetic diversity in data sets is a common challenge in genomics today. The
effects of this can be seen in diagnostic and carrier screening tools that provide more accurate and
comprehensive results for some populations than for others.
Consider these metrics about available genomic data sets: In 2009, an analysis of genome-wide
association studies found that 96% of the 1.7 million samples reviewed came from individuals of
European ancestry12. By 2021, data from the GWAS Diversity Monitor website13 indicated that the
share of GWAS samples from people with non-European ancestry had grown from that measly 4%
to just 12%.
The vast majority of historically available data supporting cystic fibrosis testing also comes from
people of European descent. The prominent historical database2 includes data from nearly 90,000
patients on pathogenic variants detected in the CFTR gene, connecting patient genotype and
phenotype. It has long been considered the gold standard on genetic data for cystic fibrosis.
Unfortunately, of the patients represented in the database, 95% have European ancestry, leaving
other populations woefully underrepresented (Figure 1).
Figure 1. Illustration of the historical variant database2 patient ethnicity compared to more realistic representation of U.S. population diversity3.
To add to the challenges in CFTR testing, there have been over 2000 CFTR mutations documented
14, with varying levels of pathogenicity. In order to provide practical cystic fibrosis
carrier screening guidance for clinical laboratories, ACMG recommended testing for 23 of the
most common mutations in 2004, based on available data at that time15.
Since then, several large-scale studies have reported the lack of coverage for diverse populations by the ACMG 23 recommended variants. One study of over 370,000 people with an ethnic distribution representative of the U.S. population found that 44% of carriers would have been missed using the ACMG 23 panel4. Another recent study of over 13,000 screened
couples with an ethnic distribution representative of the U.S. population found that 31% of at-risk couples would have been undetected using the ACMG-23, which would have resulted in missed cases of cystic fibrosis3.
Even more concerning is that most currently used targeted cystic fibrosis screening panels are at least five years old and were designed based on the historical database. Though these panels contain many more variants than the ACMG-23 panel, they still lack sufficient carrier screening coverage for the diverse U.S. population (Table 1). This is due to their design based on outdated and ethnically homogeneous data. The lack of coverage for specific ethnicities can further be seen for an on-market panel in Table 2.
Table 1. Three on-market CFTR panels with more variants detected than ACMG 23, but lacking sufficient cystic fibrosis carrier detection for the diverse U.S. population.
Table 2. Manufacturer-reported CFTR mutation detection rates of a commonly used CFTR targeted panel5. Reported coverage percentages are based on data published in 2004 or earlier, meaning coverage could be poorer if compared with more recently published, diverse datasets.
However, universal carrier screening is recommended for cystic fibrosis, which means clinicians need results that accurately represent the risk of passing on this disease for patients of all ancestries.
Large-scale population studies are adding rapidly to the information available about pathogenic variants associated with cystic fibrosis in people of diverse genetic backgrounds. Some of these studies deliberately gather data from diverse populations, such as that of the United States3,4. The rush of new, publicly-available genomic data represents significant progress in identifying relevant variants across ancestries.
Still, this information must be incorporated into carrier screening tests to make a difference for prospective parents wondering if their children would be at risk of inheriting cystic fibrosis. Clinicians ordering carrier screening tests for cystic fibrosis should reach out to their clinical lab teams to ensure that such tests have been recently updated to reflect more diverse data sources — covering not only the most common variants found in the historical database but also diverse population-specific variants identified in large-scale sequencing studies.
Spinal Muscular Atrophy (SMA) Carrier Screening
Until recently, a diagnosis of spinal muscular atrophy (SMA) was considered a death sentence. This debilitating neuromuscular disease is a leading genetic cause of death among infants. The introduction of game-changing therapies in the last few years, though, has brought new hope to families of affected individuals16.
Though these therapies have been revolutionary, they do not negate the need for carrier screening. In fact, SMA carrier screening is recommended by the American College of Obstetricians and Gynecologists (ACOG) and ACMG for all women who are pregnant or planning to become pregnant1,17. SMA has a high carrier rate of approximately 1 in 50 individuals18.
In addition, SMA screening accuracy can vary depending on ancestry. Historically, SMN1 copy number was thought to be the only information needed for SMA carrier status: if a person had two copies of SMN1 it was assumed they were on different alleles and the person could not pass on the disease to a child. Now it is known that conventional tests that report only SMN1 copy number may miss “silent carriers”, or people who carry two copies of the SMN1 gene on a single allele and no copies on the other allele (Figure 2). This silent carrier genotype can occur in any ethnicity, but it is more common among people of African ancestry9, so standard SMA screening tests may be more likely to miss carriers in this population. In fact, if reporting SMN1 copy number alone, “silent carrier” genotypes can lead to approximately 30% false negative results for African American patients9.
Figure 2. Illustration of SMN1 “silent carrier” genotype with two SMN1 copies on the same allele (center), which can allow for passing on a zero copy allele, compared to a noncarrier genotype where one SMN1 copy is present on each allele (right). One copy SMN1 carrier genotype is on left. If receiving SMN1 copy number results only, silent carriers cannot be distinguished since both center and right genotypes would yield results of 2 copies of SMN1.
Fortunately, it has been observed that certain variants (called “linked variants”) in the SMN1 gene can help predict silent carrier status. Presence of these variants can help further stratify carrier risk in all ethnicities, and even diagnose carrier status in a few19, 20 (Table 3).
Table 3. Impact of positive results for an SMN1 linked variant (c.*3+80T>G) on SMA carrier risk19, 20 when positive for two SMN1 copies, by ethnicity.
It is no longer enough to report only SMN1 copy number for carrier screening. All SMA carrier screening tests should also include detection of these linked variants to provide patients with better information on potential carrier status.
Furthermore, many commonly used tests for SMA are not as accurate or as sensitive as they should be when providing such vital clinical information. The test considered the gold standard for SMA testing is based on a cumbersome, time-consuming technology called MLPA. There has been some concern cited that this technology many not reliably distinguish certain copy numbers21. In addition, popular SMA screening technologies may not provide linked variant detection to inform carrier risk. Clinicians should check with their clinical lab teams to ensure they are using an SMA carrier screening test capable of providing this information.
Fragile X Carrier Screening
Fragile X syndrome is the most common inherited form of intellectual disability and autism, affecting approximately 1 in 4,000 males and 1 in 5,000 females in the United States22. ACOG and ACMG recommend carrier screening for this syndrome1,17,23. However, of the estimated 1 million women believed to be Fragile X carriers in the U.S.22, the majority may be unaware of their carrier status.
Fragile X syndrome is caused by a CGG repeat expansion in the FMR1 gene on the X chromosome. Different counts of the repeat have different phenotypic outcomes (Table 4). Healthy individuals typically have fewer than 45 CGG repeats, while those who may be unaffected carriers or have associated disorders such as Fragile X-Associated Primary Ovarian Insufficiency (FXPOI) generally have between 55 and 200 repeats. Individuals with at least 200 repeats are diagnosed with Fragile X syndrome.
Table 4. Clinical categories and carrier status of mutations in the FMR1 gene24,25,26.
Abbreviations: FXS, fragile X syndrome; FXTAS, fragile X-associated tremor/ataxia syndrome; FXPOI, fragile X-associated primary ovarian insufficiency.
For any genetic testing platform, obtaining an accurate count of so many repeated DNA triplets is a serious challenge. Additionally, many analysis platforms cannot accurately interrogate the region due to the technical challenges of assessing GC-rich DNA content. Therefore, it is important that fragile X carrier screening is being performed with robust technology that can analyze extreme GC content and provide accurate counts of CGG repeats in order to identify women at risk of having a child with fragile X.
Figure 3. AGG interruption quantification stratifies risk of expansion to full mutation in the next generation10,11,27. Risk by CGG repeat number only represented by blue line; 0 AGG interruptions detected, orange line; 1 AGG interruption detected, black line; 2 AGG interruptions detected, teal line. A 70 CGG patient example is illustrated where 21% risk is reported with CGG repeat-only information, which can be further stratified to range from 50% risk when 0 AGG interruptions (orange arrow) are detected to near 0% risk when 2 AGG interruptions are detected (teal arrow).
Beyond CGG repeat counts, clinicians should also be provided information on AGG interruptions for women with 50 – 90 CGG repeats. In this select group, the number of AGG interruptions stratifies risk of expansion to a full mutation in offspring beyond that provided by CGG repeats alone (Figure 3). For example, for a woman with a 70 CGG repeat allele, risk of expansion to a full mutation can range from ~0% to 50% depending on the number of AGG interruptions. Information provided by the AGG interruption status can greatly affect reproductive decisions; thus, clinicians should work with their clinical lab partners to ensure they are providing this information for select patients.
Conclusion
As insight into inherited diseases grows and diagnostic technology advances, providing all patients with accurate and relevant information must remain the central goal. Thankfully, due to the diligence of many scientists and clinicians, better tests have been developed for cystic fibrosis, spinal muscular atrophy, and fragile X carrier screening. As guidelines have shifted to pan-ethnic carrier screening, it is vital that clinicians and their clinical lab partners are aware of disparities and gaps in genetic information from often-used, yet outdated tests, and that they strive to provide more comprehensive and reliable information for all patients.
References
1. Gregg AR, et al. Genet Med. 2021 Oct;23(10):1793-1806.
2. Sosnay et al Nat Genet. 2013 Oct; 45(10): 1160–1167
3. Beauchamp KA, et al. Genet Med. 2019; 21(11): 2569–2576.
4. Westemeyer M, et al. Genet Med. 2020 Aug;22(8):1320-1328.
5. Luminex xTAG® Cystic Fibrosis (CFTR) 60 kit v2 product brochure.
6. McGarry ME, et al. Pediatr Pulmonol. 2019 Nov;54 (Suppl 3):S74-S83.
7. Watts KD, et al. J Genet Couns. 2012 Oct;21(5):671-5. doi: 10.1007/s10897-012-9481-2.
8. Neemuchwala F, et al. Case Rep Pediat. 2018 Jul 2;2018:7217326.
9. Hendrickson BC, et al. J Med Genet. 2009 Sep;46(9):641-4.
10. Nolin SL, et al. Genet Med. 2015 May;17(5):358-64.
11. Nolan SL, et al. Am J Med Genet A. 2013 Apr;161A(4):771-8.
12. Need AC and Goldstein DB. Trends Genet. 2009 Nov;25(11):489-94.
13. Mills MC and Rahal C. GWAS Diversity Monitor. 2021. https://gwasdiversitymonitor.com/
14. Veit G, et al. Mol Biol Cell. 2016 Feb 1; 27(3): 424–433.
15. Watson MS, et al. Genet Med. Sep-Oct 2004;6(5):387-91.
16. Nance JR. Spinal Muscular Atrophy. Continuum (Minneap Minn). 2020 Oct;26(5):1348-1368.
17. Rink B, et al. ACOG Opinion No 691. March 2017. Reaffirmed 2020.
18. Cure SMA. 2021. https://www.curesma.org/carriers-of-sma/
19. Luo, et al. Genet Med. 2014 Feb;16(2):149-56.
20. Alías L, et al. Eur J Hum Genet. 2018 Oct;26(10):1554-1557.
21. Schorling DC, et al. Neurology. 2019 Aug 6;93(6):267-269.
22. National Fragile X Foundation. 2021. https://fragilex.org/
23. Sherman S, et al. Genet Med. 2005 Oct;7(8):584-7.
24. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 300623. 2019. https://omim.org/
25. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 300624. 2019. https://omim.org/
26. Allen EG, et al. Genet Med. 2021 Sep;23(9):1648-1655.
27. Villate O, et al. Front Mol Biosci. 2020 Jul 14;7:135.